Escuchar esta nota
Detenido
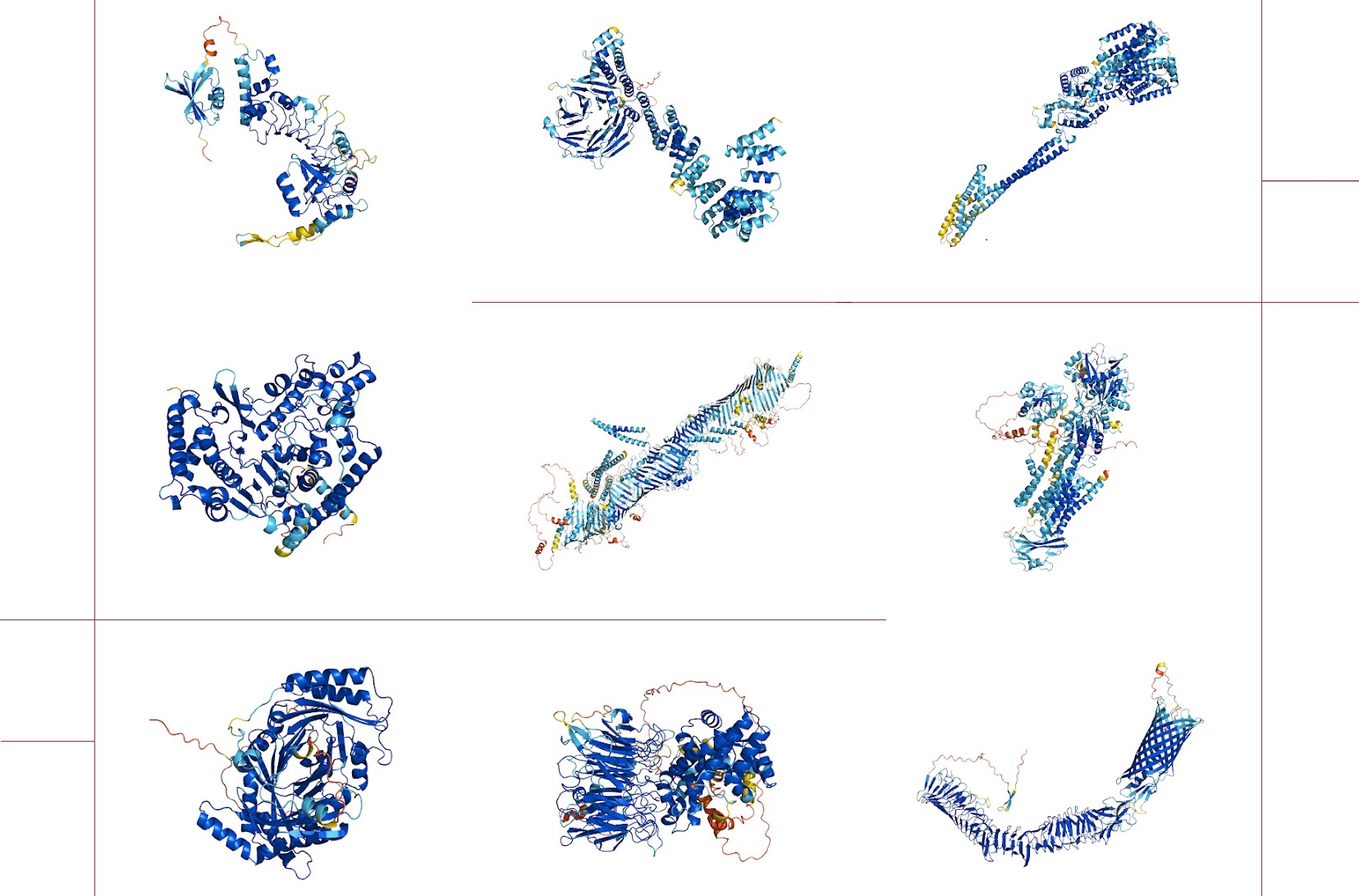
La Base de Datos de Estructura de Proteínas AlphaFold da a conocer a la comunidad científica más de 350 000 predicciones de estructuras de proteínas, incluido todo el proteoma humano; con ello, pretende promover la investigación en muchos campos para comprender el papel de estos componentes básicos de la vida. La suma de matemáticas y bioinformática ha sido clave para lograr este hito científico, subrayan, por otra parte, las científicas del CSIC Nuria Campillo y Carmen Fernández.
DeepMind ha anunciado en días recientes su colaboración con el Laboratorio Europeo de Biología Molecular (EMBL), el principal laboratorio europeo en ciencias de la vida, para proporcionar de manera libre y abierta a la comunidad científica la base de datos de los modelos de predicciones de las estructuras del proteoma humano (el conjunto completo de proteínas codificadas por el genoma humano) más completa y precisa hasta la fecha.
Esto incluirá alrededor de 20 000 proteínas expresadas por el genoma humano. La base de datos y el sistema de inteligencia artificial brindan a los biólogos estructurales nuevas y poderosas herramientas para examinar la estructura tridimensional de las proteínas, y ofrecen un tesoro de datos que podría abrir el camino a futuros avances y presagiar una nueva era para la biología basada en la inteligencia artificial.
En diciembre de 2020, los organizadores de la evaluación comparativa Critical Assessment of Protein Structure Prediction (CASP) reconocieron AlphaFold como una solución al gran desafío de más de 50 años de predecir la estructura de proteínas, lo que significó un logro asombroso en el campo.
La base de datos de estructura de proteínas AlphaFold (AlphaFold Protein Structure Database) se basa en esta innovación y en los descubrimientos de generaciones de científicos y científicas, desde los pioneros y las pioneras de la cristalografía y el análisis de estructura de las proteínas, hasta los miles de especialistas en predicción y biólogos y biólogas estructurales que han pasado años experimentando con proteínas desde entonces y que han compartido sus resultados de forma abierta.
La base de datos amplía drásticamente el conocimiento acumulado sobre las estructuras de proteínas, más que duplicando el número de estructuras de proteínas humanas con predicciones de alta precisión disponibles para los investigadores. Avanzar en la comprensión de estos componentes básicos de la vida, que sustentan los procesos biológicos en todos los seres vivos, permitirá a los investigadores de una gran variedad de campos acelerar su trabajo.
Hace unas semanas, se publicó en la revista Nature la metodología de la última e innovadora versión de AlphaFold, el sofisticado sistema de inteligencia artificial anunciado en diciembre pasado que impulsa estas predicciones de estructura, y su código fuente abierto. El anuncio de ahora coincide con un segundo artículo de Nature que proporciona la imagen más completa de las proteínas que componen el proteoma humano, y la publicación de las proteínas de 20 organismos adicionales que son importantes para la investigación biológica.
“Nuestro objetivo en DeepMind siempre ha sido construir inteligencia artificial y utilizarla como una herramienta para ayudar a acelerar el ritmo del descubrimiento científico, y mejorar así el conocimiento del mundo que nos rodea”, explica el fundador y director ejecutivo de DeepMind, Demis Hassabis.
“Hemos utilizado AlphaFold para generar la imagen más completa y precisa del proteoma humano. Creemos que esta es la contribución más significativa que ha hecho la inteligencia artificial al avance del conocimiento científico hasta la fecha, y es un gran ejemplo de los tipos de beneficios que la inteligencia artificial puede aportar a la sociedad”, ha añadido.
Una ayuda a los científicos para acelerar sus descubrimientos
La capacidad de predecir computacionalmente la forma de una proteína a partir de su secuencia de aminoácidos, en lugar de tener que determinarla experimentalmente con técnicas minuciosas, laboriosas, y a menudo costosas, ya está ayudando a los científicos a lograr en meses lo que antes requería años de trabajo.
“La base de datos AlphaFold es un ejemplo perfecto del círculo virtuoso de la ciencia abierta”, explica la directora general del EMBL, Edith Heard. “AlphaFold ha sido entrenado utilizando datos de recursos públicos creados por la comunidad científica, por lo que tiene sentido que sus predicciones sean públicas. Compartir las predicciones de AlphaFold de forma abierta y gratuita permitirá a los investigadores de todo el mundo obtener nuevos conocimientos e impulsar nuevos descubrimientos. Creo que AlphaFold es una verdadera revolución para las ciencias de la vida, así como fue la genómica hace varias décadas y estoy muy orgullosa de que el EMBL haya podido ayudar a DeepMind a permitir el acceso abierto a este recurso extraordinario”, añade.
AlphaFold ya está siendo utilizado por socios como la Iniciativa de Medicamentos para Enfermedades Desatendidas (DNDi, por sus siglas en inglés), que ha avanzado en su investigación sobre curas que salvan vidas para enfermedades que afectan de manera desproporcionada a las zonas más empobrecidas del mundo, o el Centro de Innovación Enzimática (CEI) que utiliza AlphaFold para ayudar a diseñar enzimas más rápidas para reciclar algunos de los plásticos más contaminantes de un solo uso.
AlphaFold ha ayudado a acelerar la investigación de aquellos científicos y científicas que trabajan en la determinación experimental de la estructura de las proteínas. Por ejemplo, un equipo de la Universidad de Colorado en Boulder utiliza las predicciones de AlphaFold para estudiar la resistencia a los antibióticos, mientras que un grupo de la Universidad de California en San Francisco las ha utilizado para estudiar la biología del SARS-CoV-2.
La base de datos de estructura de proteínas AlphaFold
La base de datos de estructura de proteínas AlphaFold está basada en muchas contribuciones de la comunidad científica internacional, así como en las refinadas innovaciones algorítmicas de AlphaFold y en las décadas de experiencia del Instituto Europeo de Bioinformática del EMBL (EMBL-EBI) compartiendo datos biológicos mundiales. DeepMind y el EMBL-EBI están dando libre acceso a las predicciones de AlphaFold para que cualquiera pueda usar el sistema con el fin de permitir y acelerar la investigación y explorar nuevas vías de conocimiento científico.
“Este será uno de los conjuntos de datos más importantes desde el mapa del Genoma Humano”, recalca el Director General Adjunto del EMBL y el director del EMBL-EBI, Ewan Birney. “Hacer que las predicciones de AlphaFold sean accesibles a la comunidad científica internacional abre muchas nuevas vías de investigación, desde enfermedades desatendidas hasta nuevas enzimas para la biotecnología y mucho más. Esta es una nueva y gran herramienta científica, que complementa las tecnologías existentes y nos permitirá ampliar los límites de nuestra comprensión del mundo”.
Entre las primeras más de 350 000 estructuras publicadas en la base de datos, además del proteoma humano, están las proteínas de 20 organismos biológicamente significativos como E. coli, la mosca de la fruta, el ratón, el pez cebra, el parásito de la malaria y las bacterias de la tuberculosis. Se han realizado muchas investigaciones importantes sobre estos organismos, y tener estas estructuras a disposición permitirá a muchos investigadores de campos muy diferentes, desde la neurociencia hasta la medicina, acelerar su trabajo.
La base de datos y el sistema serán actualizados periódicamente a medida que se continúe invirtiendo en mejoras futuras de AlphaFold, y en los próximos meses se planea expandir enormemente la cobertura a casi todas las proteínas secuenciadas conocidas por la ciencia: más de 100 millones de estructuras que incluyen la mayoría de UniProt, la base de datos referencia.
Referencia: Kathryn Tunyasuvunakool et al.
“Highly accurate protein structure prediction for the human proteome” – Nature.
◾◾◾
La suma de matemáticas y bioinformática desentraña las piezas básicas de los seres humanos
Núria Campillo / Carmen Fernández
Hace más de 50 años, científicos de la talla de los premiados con el Nobel, Kendrew, Perutz, o Hodgkin, estaban metidos de lleno en el estudio de las proteínas, el milagro de la vida, mediante cristalografía de rayos X. El plegamiento, no sólo de las proteínas, sino de las macromoléculas en general (enzimas y receptores), junto con poder discernir su estructura tridimensional, ha sido uno de los desafíos científicos de las últimas décadas.

Diferentes disciplinas, como la ya mencionada cristalografía, la criomicroscopía electrónica, la resonancia magnética nuclear (RMN), la bioinformática y la modelización in silico, han conseguido increíbles logros en este sentido. Sin embargo, el trabajo de resolver la estructura de estas macromoléculas sigue siendo complicado y costoso.
Y este es un desafío importante, porque las macromoléculas responsables de la vida, como las proteínas, son elementos clave para el buen funcionamiento de nuestro organismo, estando involucradas en prácticamente todos los procesos biológicos que tienen lugar en nuestras células. Poder conocer la estructura de las proteínas posibilita, de una forma más sencilla, determinar su función y, por lo tanto, poder entender y solucionar problemas cuando alguna de ellas no funciona como debería.
Así contribuye la inteligencia artificial
La inteligencia artificial ha llegado por la puerta grande a este campo, como en otras muchas áreas del conocimiento, para ayudar a resolver, por ejemplo, la predicción de la estructura 3D de una manera más rápida. Nos va a permitir poder predecir la forma y, como hemos dicho antes, el funcionamiento que tienen las proteínas (un conocimiento indispensable para el desarrollo de tratamientos eficaces para cualquier enfermedad) o las enzimas (llaves maestras en muchos procesos tanto biológicos como industriales), entre otras macromoléculas.
Recientemente se ha conocido el ganador del concurso científico CASP 14 (del inglés Critical Assessment of Protein Structure Prediction). A este concurso internacional concurren cientos de grupos de investigación para presentar el desarrollo de sus softwares o aproximaciones para predecir la estructura de proteínas a partir de sus secuencias de aminoácidos. Podríamos decir que es el escaparate de los desarrollos teóricos más prometedores para la elucidación de la estructura 3D de las proteínas. El ganador, sin ningún tipo de duda, ha sido el algoritmo AlphaFold, desarrollado por la empresa DeepMind (perteneciente a Google).
La mayoría de las predicciones de AlphaFold para la estructura 3D de las proteínas resueltas son muy similares a las que se obtienen a partir de las técnicas experimentales. AlphaFold consiguió una puntuación de casi 90 sobre 100 en un test que mide la semejanza estructural entre la determinada experimentalmente y la predicha por el algoritmo (por encima de 90 se considera como un nivel de predicción total). Nunca antes se había alcanzado una puntuación por encima de 60 en este test.
Los impresionantes resultados han sido posibles gracias a la sinergia de diferentes disciplinas: matemáticas, bioinformática, biología… El sistema informático parte de la premisa de que la estructura plegada tridimensional de una macromolécula (enzimas, proteínas, receptores) se asemeja a un grafo espacial, donde los aminoácidos son los nodos y las aristas del grafo conectan dichos aminoácidos.
Partiendo de esta idea y utilizando el conocimiento derivado del alineamiento de cientos de miles de secuencias de proteínas de estructura 3D conocidas, AlphaFold utiliza la metodología del aprendizaje profundo (deep learning) para aprender las características físicas, geométricas y evolutivas de esos datos de entrada. Los desarrolladores han hecho un buen trabajo enseñando a AlphaFold a extraer información de lo ya conocido para después aplicar dicho conocimiento ante una nueva situación: determinar la estructura de proteínas para las que solo conocemos sus secuencias de aminoácidos, y hacerlo con una precisión y rapidez que no están al alcance de los métodos tradicionales.
Las aplicaciones no se han hecho esperar. DeepMind y el Laboratorio Europeo de Biología Molecular (EMBL), publicaron la semana pasada más de 350 000 nuevas estructuras, incluyendo, no sólo unas 20 000 proteínas humanas, sino también proteínas de otros 20 organismos, como las de las bacterias E. Coli, la causante de la tuberculosis, entre otras, o las de los ratones que se utilizan para los experimentos en los laboratorios. Y ya hay organizaciones, como la Iniciativa Medicamentos para Enfermedades Olvidadas (DNDi) que están rastreando esta nueva base de datos de proteínas en busca de nuevos tratamientos.
Evidentemente todavía queda mucho camino por andar y numerosos desafíos a los que enfrentarnos, pero AlphaFold ya ha abierto las puertas a abordar un gran reto: ahondar en el secreto de la vida.



